微观下的生物智能分析 | 计算机与医学图像领域顶级会议 MICCAI 2022收录西湖大学杨林实验室最新成果3项
MICCAI是由国际医学图像计算和计算机辅助干预协会(Medical Image Computing and Computer Assisted Intervention Society) 举办,跨医学影像计算(MIC)和计算机辅助介入 (CAI) 两个领域的综合性学术会议,是该领域的顶级会议。近日,杨林团队的3项科研成果被MICCAI收录,在染色体拉直技术、病理图像泛化性及细胞识别技术上取得了创新性进展。
1.ChrSNet: Chromosome Straightening using Self-attention Guided Networks
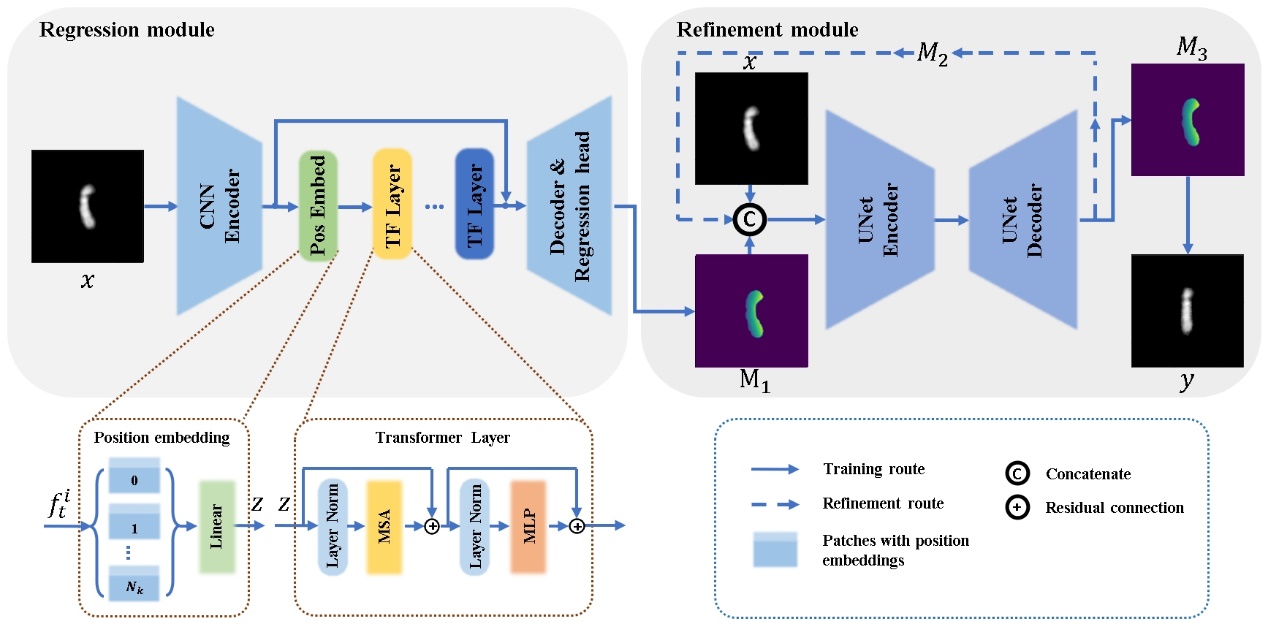
染色体核型分析是染色体分类和染色体异常诊断的重要方法。在染色体显微镜图像中,由于染色体非刚性的特点,染色体往往呈弯曲形态。这种弯曲形态会影响细胞遗传学家分析染色体核型的效率和准确度。为了解决这个问题,杨林团队提出了一个自注意力引导框架来消除染色体的弯曲。所提出的框架包括回归模块和细节优化模块,以用于还原并修正染色体的形态学特征和结构特征。实验结果表明,杨林团队提出的方法可以有效地拉直弯曲染色体,并保持染色体的条带特征和长度信息。
2.Benchmarking the Robustness of Deep Neural Networks to Common Corruptions in Digital Pathology
数字病理图像制片过程复杂,容易产生严重的图像扰动,导致目前大部分数字病理智能分析算法在多中心验证中性能下降明显。杨林团队在该篇论文成果中归纳总结出9种常见的图像扰动(JPEG压缩失真、Pixelate拉伸失真、失焦模糊、运动模糊、亮度变化、饱和度变化、色相变化、图像污迹、气泡(如图2))并且仿真出这些扰动。通过将扰动加入到现有数据集的验证集中,同时设计在扰动下评估模型准确率和置信度的指标,构建了病理图像鲁棒性验证基准。杨林团队在两个多中心、大规模的病理图像数据集上进行验证,揭示了当前深度模型在扰动下脆弱的鲁棒性。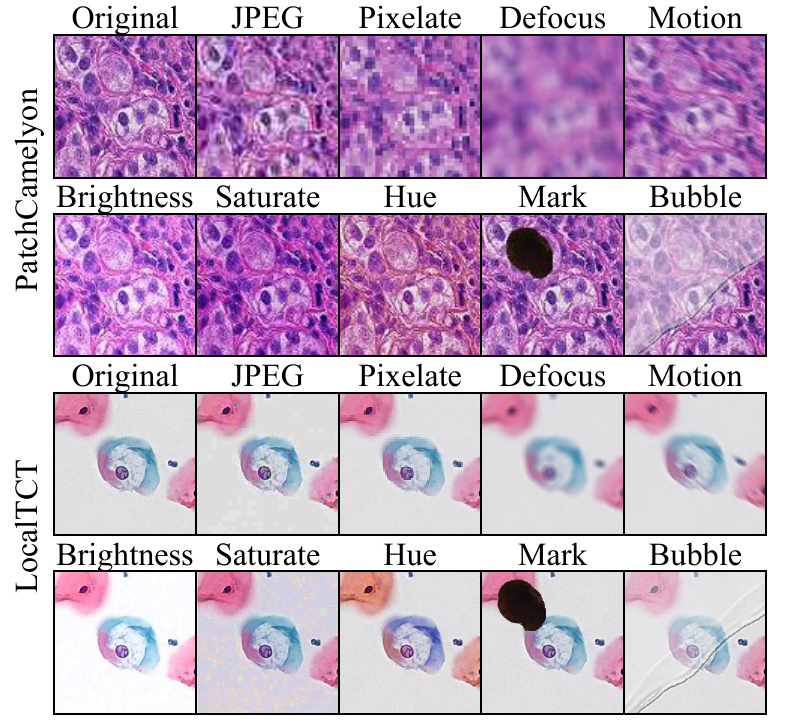
3.End-to-End cell recognition by point annotation
细胞识别对于免疫组化染色图像的定量分析具有重要意义。通常的弱监督建模方法是先预测细胞的概率密度图,再采用局部最大值检测的后处理方法定位细胞。然而,由于细胞的密度在不同切片及同一切片的不同区域间存在较大差异,无法找到一套通用的后处理参数, 因此,这些基于密度图的模型的性能在密集细胞识别的场景下会受到极大的限制。
在此项工作中,杨林团队提出了一个端到端的细胞识别框架,对预设的锚点直接进行回归和分类。为了构建更具判别性的特征,将图像的底层特征(如梯度利于细胞定位、颜色利于细胞分类)与高级语义特征相结合。对比现有的细胞识别方法,杨林团队的方法具有更高的准确性和推断效率,在协助免疫组化染色图像分析方面具有很高的潜力。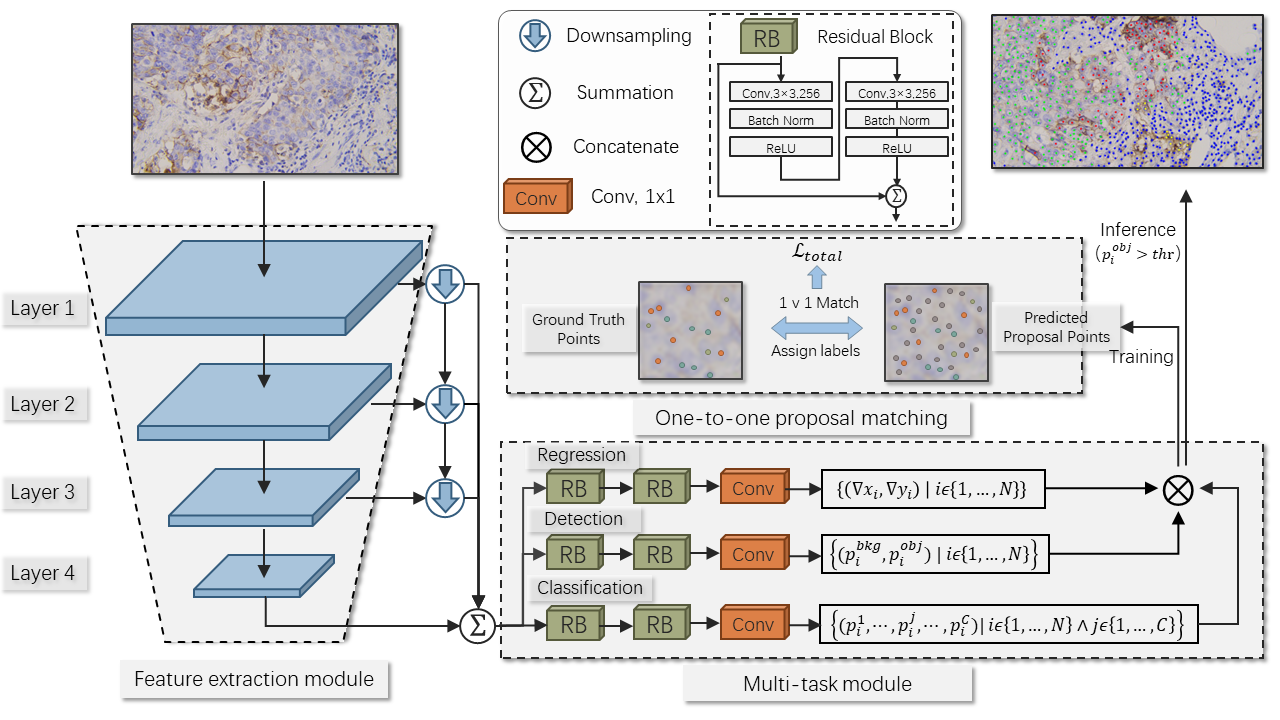
*上述三项研究均在西湖大学杨林教授(通讯作者)的指导下完成,其中,”ChrSNet: Chromosome Straightening using Self-attention Guided Networks”由杨林实验室博士后郑孙易和2021级博士生李竞雄共同完成(共同一作); ”Benchmarking the Robustness of Deep Neural Networks to Common Corruptions in Digital Pathology” 由2021级博士生章云龙和科研助理孙宇轩(2022级博士生)共同完成(共同一作); ”End-to-End cell recognition by point annotation” 由实验室访问学生水忠益(2022级博士生)和2020级博士生张士川共同完成(共同一作)。
1.ChrSNet: Chromosome Straightening using Self-attention Guided Networks
染色体核型分析是染色体分类和染色体异常诊断的重要方法。在染色体显微镜图像中,由于染色体非刚性的特点,染色体往往呈弯曲形态。这种弯曲形态会影响细胞遗传学家分析染色体核型的效率和准确度。为了解决这个问题,杨林团队提出了一个自注意力引导框架来消除染色体的弯曲。所提出的框架包括回归模块和细节优化模块,以用于还原并修正染色体的形态学特征和结构特征。实验结果表明,杨林团队提出的方法可以有效地拉直弯曲染色体,并保持染色体的条带特征和长度信息。
图1:染色体拉直模型结构
原文链接:https://arxiv.org/pdf/2207.00147.pdf
2.Benchmarking the Robustness of Deep Neural Networks to Common Corruptions in Digital Pathology
数字病理图像制片过程复杂,容易产生严重的图像扰动,导致目前大部分数字病理智能分析算法在多中心验证中性能下降明显。杨林团队在该篇论文成果中归纳总结出9种常见的图像扰动(JPEG压缩失真、Pixelate拉伸失真、失焦模糊、运动模糊、亮度变化、饱和度变化、色相变化、图像污迹、气泡(如图2))并且仿真出这些扰动。通过将扰动加入到现有数据集的验证集中,同时设计在扰动下评估模型准确率和置信度的指标,构建了病理图像鲁棒性验证基准。杨林团队在两个多中心、大规模的病理图像数据集上进行验证,揭示了当前深度模型在扰动下脆弱的鲁棒性。
图2:添加9种扰动后的两张示例
原文链接:https://arxiv.org/pdf/2206.14973.pdf
3.End-to-End cell recognition by point annotation
细胞识别对于免疫组化染色图像的定量分析具有重要意义。通常的弱监督建模方法是先预测细胞的概率密度图,再采用局部最大值检测的后处理方法定位细胞。然而,由于细胞的密度在不同切片及同一切片的不同区域间存在较大差异,无法找到一套通用的后处理参数, 因此,这些基于密度图的模型的性能在密集细胞识别的场景下会受到极大的限制。
在此项工作中,杨林团队提出了一个端到端的细胞识别框架,对预设的锚点直接进行回归和分类。为了构建更具判别性的特征,将图像的底层特征(如梯度利于细胞定位、颜色利于细胞分类)与高级语义特征相结合。对比现有的细胞识别方法,杨林团队的方法具有更高的准确性和推断效率,在协助免疫组化染色图像分析方面具有很高的潜力。
图3:细胞识别模型训练及推断流程图
原文链接:https://arxiv.org/pdf/2207.00176.pdf
*上述三项研究均在西湖大学杨林教授(通讯作者)的指导下完成,其中,”ChrSNet: Chromosome Straightening using Self-attention Guided Networks”由杨林实验室博士后郑孙易和2021级博士生李竞雄共同完成(共同一作); ”Benchmarking the Robustness of Deep Neural Networks to Common Corruptions in Digital Pathology” 由2021级博士生章云龙和科研助理孙宇轩(2022级博士生)共同完成(共同一作); ”End-to-End cell recognition by point annotation” 由实验室访问学生水忠益(2022级博士生)和2020级博士生张士川共同完成(共同一作)。